Iranian Journal of Catalysis (IJC) publishes experimental and theoretical research results of outstanding significance in the field of catalysts and their application in organic, inorganic, bioorganic, analytical, polymer, and other branches of chemistry. The scope of IJC includes homogeneous and heterogeneous catalysis, catalytic reactions, computational catalysis, synthesis and catalytic function of novel inorganic solids and complexes, enzymatic catalysis, and spectroscopic methods for structural characterization.
Journal DOI: 10.57647/IJC
IJC is an open access, peer-reviewed journal (double-blinded), meaning that all interested readers can freely access the journal online at http://ijc.shahreza.iau.ir without needing a subscription. Manuscripts accepted for publication will not be subject to any page charges, color charges, or article processing charges and immediately appear online, followed by a printed hard copy.
In Addition, the “Iranian Journal of Catalysis” has been indexed on the “Web of Science“.

The “Iranian Journal of Catalysis” Now have included in Journal Impact Factor List of “Clarivate”, 2022. (JCR)
The “Iranian Journal of Catalysis” has become a member of COPE, the Committee on Publication Ethics, since January 2013.

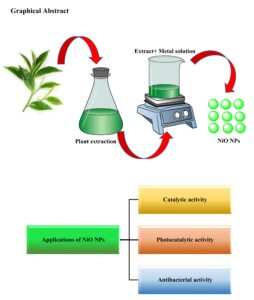
In the past decade, numerous longitudinal studies have explored green chemistry and its applications in nanoparticle synthesis due to the toxicity associated with traditional methods. Among the various techniques for nanoparticle synthesis, the use of plant extracts in green synthesis has recently gained significant popularity. Green methods are particularly suitable for large-scale nanoparticle synthesis, offering […]
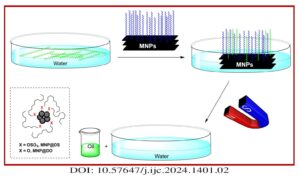
Magnetic nanoparticles (MNPs) were treated with dodecyl sulfate (DS) and dodecanol (DO) to enhance their affinity for lipids. Various methods, such as X-ray powder diffraction (XRD), scanning electron microscopy (SEM), vibrating sample magnetometery (VSM), thermal gravimetric analysis (TGA), and FTIR spectroscopy were employed to analyze and characterize these modified MNPs. The functionalized MNPs were employed […]
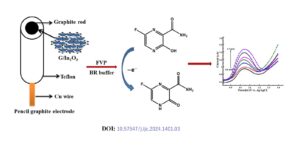
Favipiravir (FVP) was one of the promising medications for the treatment of Covid-19 patients. There are limited studies on the electrochemical performance of FVP. Hence it is interesting to study the electrochemical detection of FVP by cyclic voltammetry (CV), differential pulse voltammetry (DPV) and chronoamperometry. The G/In2O3 nanocatalyst was prepared by precipitation method and applied […]

, since 2013.")


")

